Create submit scripts
Create submit script
A submit script is a shell script with which a calculation job is transferred to the Linux cluster. Comments start with the # character(# at the beginning of the line). Lines beginning with #SBATCH are interpreted by SLURM as parameters for resource requests and other submit script options. While these instructions only deal with the creation of a submit script, the next chapter, Starting programs in batch mode, will show you how to start it and display the results.
The following table lists the most common parameters with a brief explanation.
sbatch
| Command | Description |
|---|---|
| #SBATCH --partition=public | All computers should come from the "public" partition. More on this under "Batch operation with SLURM". Default: public |
| #SBATCH --nodes=2 | 2 computers are required |
| #SBATCH --tasks-per-node=4 | 4 of the CPUs are required by each computer |
| #SBATCH --mem=1000 | 1000 MB main memory per computer is required (21000 MB * maximum number of allocated CPUs). Cannot be combined with "--mem-per-cpu"! |
| #SBATCH --mem-per-cpu=100 | At least 100 MB main memory is required per CPU (21000 MB maximum), cannot be combined with "--mem"! |
| #SBATCH --time=0:01:00 | The job will require a maximum of 1 minute. If the value is exceeded, the job is aborted. The maximum is parition-dependent. Format hh:mm:ss |
| #SBATCH --constraint=12cores | Nodes for which the "12 processor cores" feature is entered. With sinfo --format="%20P %.40f" a list with all defined features is displayed. |
| #SBATCH --exclusive | Computer systems are required exclusively, no sharing with other jobs. Use sparingly. |
| #SBATCH --nodelist=its-cs104 | The machine its-cs104 is required. further example value: its-cs[266,271-274,302] |
| #SBATCH -output=filename | Program output should be written to the specified file. |
| #SBATCH -error=filename | Error output should be written to the specified file. |
| #SBATCH --mail-type=BEGIN|END|FAIL|REQUEUE|ALL | E-mail at start, end, abort, requeue or all status changes of the job. Use only one option per line. |
| #SBATCH --mail-user=[your email address] | E-mail address for notifications. |
| #SBATCH --job-name="MyJob" | Give the job a name. Several jobs can have the same name. For example, the job name can be used in the submit script with %N to name the .out file. You can also use scancel <jobname> to cancel all jobs with the specified job name at the same time. |
In the following example, a node in the partition public with 100 MB main memory is allocated for 5 minutes. At the end of the script, a program is usually started. In our case, only the name of the server on which the program was executed is output as an example.
| Example 1: myscript.sh | |
|---|---|
| #!/bin/bash | |
| #SBATCH --partition=public | # Partition public |
| #SBATCH --nodes=1 | # 1 node is required |
| #SBATCH --tasks-per-node=1 | # number of tasks |
| #SBATCH --mem=100 | # 100 MB main memory |
| #SBATCH --time=0:05:00 | # max. runtime 5 min |
| #SBATCH --output=slurm.%j.out | # file for stdout |
| #SBATCH --error=slurm.%j.err | # File for stderr # (%N: node name, %j: job no.) |
| #SBATCH --mail-type=END | # Notification at the end |
| #SBATCH --mail-type=FAIL | # Notification on error |
| #SBATCH --mail-user=user@host.de | # EMail address |
| echo "Hello from `hostname`" | # Execute program |
You can now run this submit script in batch mode for testing. The following should happen:
- The job number appears on the console
- The job waits in a queue until it is processed
- The output file slurm.12345678.out is stored in the directory of the submit script
- You receive an email that the job has been completed
If you output the file e.g. with cut slurm.12345678.out, the name of the node that executed the job should appear in it.
You can also specify the same file for the output of the program and the output of the error messages. If your program does not have many outputs anyway, this may be clearer.
Submit Script Generator
With the script generator it is possible to generate submit scripts suitable for the Linux Cluster Kassel. You can then extend the default by loading modules and starting your program.
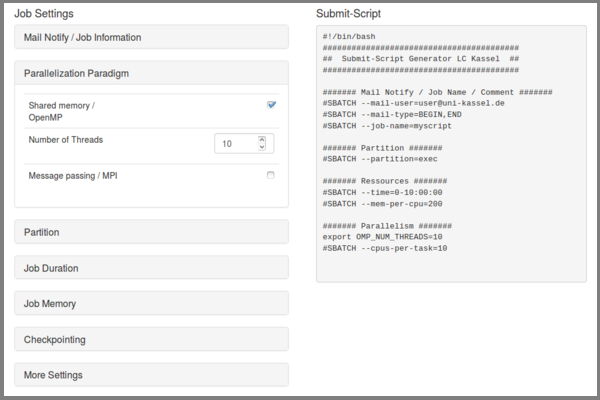
In the left-hand area there are expandable elements in which special resource requirements or job information can be specified. The code generated in the right-hand area can be inserted into a shell script using copy&paste. It is of course necessary to extend the script with the command for calling your own program or similar.
Parallel jobs
There are various options for creating parallel jobs whose processes are executed simultaneously:
- MPI application
- Multithreaded program with OpenMP
- Multiple instances of a single-threaded program
Tasks and processes
For the SLURM workload manager, a task is a process. A multi-process program consists of several tasks, whereas a multithreaded program consists of only one task that can use several CPUs.
The --ntasks option creates a number of tasks in a multi-process program. CPUs, on the other hand, are requested for multithreaded programs with --cpus-per-task. Tasks cannot be distributed to several nodes of a cluster. The --cpu-per-task option would therefore allocate several CPUs to one and the same node. In contrast, --ntasks would request several CPUs on different nodes.
Submit script for OpenMP
Here, four cores are allocated for one task. The script hello_openmp.c is compiled with gcc -fopenmp hello_openmp.c -o hello_openmp and called at the end of the submit script with ./hello_openmp:
Example 3: myscript_openmp.sh |
|---|
#!/bin/bash |
The output of the program is
Hello World from thread 0 |
Submit script for MPI
In the following example, two compute nodes are allocated 100 MB per CPU for ten minutes in the exec partition. Two processes are executed on each of the two nodes. The program hello_mpi.c was previously compiled with mpicc hello_mpi.c -o hello_mpi (compilers can be changed using the module system on the cluster) and started with mpirun at the end of the script:
Example 2: myscript_mpi.sh |
|---|
#!/bin/bash |
The output of the program could look like this:
Hello world from processor its-cs240.its.uni-kassel.de, rank 1 out of 4 processors |
Tip: You can also obtain the output of all nodes involved by using the command echo "Machine list:" $SLURM_NODELIST directly in the submit script.
Submit script for OpenMP with MPI (hybrid)
If a program uses OpenMP and MPI, both must of course be defined in the SubmitScript. The program hello_hybrid.c is compiled with mpicc -fopenmp hello_hybrid.c -o hello_hybrid. In the following example, 2 nodes (MPI processes) are requested for the program, each of which in turn uses 4 threads (OpenMP threads).
Example 4: hybrid_submitscript.sh |
|---|
#!/bin/bash |
The output of the program is:
Used nodes: its-cs[300,302] |
Job Array
With the help of the job array, it is possible to execute a large number of jobs of the same processes with different parameters or transfer files. After executing the submit script, a certain number of "Array Tasks" are created in which the variable SLURM_ARRAY_TASK_ID has a unique value. The value is specified via the index(#SBATCH --array=<index>).
In the following script, ten array tasks with the indices 10, 20, ... , 100 are created. At the end of each array task, the variable $SLURM_ARRAY_TASK_ID, which has the index as its value, is passed as a parameter to the program to be executed:
Example 4: myscript_job_array.sh |
|---|
#!/bin/bash |
Master/Slave
Master/slave operation can also be realized with SLURM. SLURM creates several tasks to which different commands can be assigned with a further configuration file:
Example 5-1: multi.conf |
|---|
0 echo 'I am the Master' |
Example 5-2: myscript_master_slave.sh |
|---|
#!/bin/bash |
The output of the program can be found in the defined output file:
I am the Master |